Guía profesional para analizar las propiedades térmicas de los materiales
¿Cómo responden los materiales al calor? Comprender las propiedades térmicas es crucial para las aplicaciones de ingeniería, ya que influye en todos los aspectos, desde la selección de materiales hasta las predicciones de rendimiento. Este artículo explora varios métodos utilizados en el análisis térmico, como el análisis termogravimétrico (TGA) y la calorimetría diferencial de barrido (DSC), y sus aplicaciones prácticas. Los lectores comprenderán cómo estas técnicas ayudan a predecir el comportamiento de los materiales ante los cambios de temperatura, mejorando la fiabilidad y seguridad de los proyectos de ingeniería.
La esencia del análisis térmico es el análisis de la temperatura.
La tecnología del análisis térmico consiste en medir el cambio de las propiedades físicas de las sustancias con la temperatura bajo el control de la temperatura programada (es decir, velocidad constante de aumento de la temperatura, velocidad constante de descenso de la temperatura, temperatura constante o aumento escalonado de la temperatura, etc.), que se utiliza para estudiar el cambio de los parámetros físicos, como los térmicos, mecánicos, acústicos, ópticos, eléctricos, magnéticos, etc. de las sustancias a una temperatura específica, a saber, P=f (T).
El cambio de temperatura se diseña según una regla determinada, es decir, la temperatura de control del programa: T=(t), por lo que su propiedad es a la vez función de la temperatura y del tiempo: P=f (T, t).
Importancia del análisis térmico de materiales
Se utiliza ampliamente para caracterizar las propiedades térmicas, físicas y mecánicas y la estabilidad de los materiales.
Tiene una gran importancia práctica para la investigación y el desarrollo de materiales y el control de calidad en la producción.
Breve historia del análisis térmico
Interpretación de los métodos habituales de análisis térmico
Según la inducción y clasificación de la Asociación Internacional de Análisis Térmico (ICTA), los métodos de análisis térmico actuales se dividen en nueve categorías y diecisiete tipos.
Entre los métodos de análisis térmico más utilizados se encuentran el análisis termogravimétrico (TG), la calorimetría diferencial de barrido (DSC), la estática termomecánica (TMA), análisis termomecánico dinámico (DMTA), análisis dieléctrico dinámico (DETA), etc.
Son funciones de medición del peso del material, el calor, el tamaño, el módulo, la conformidad, la constante dieléctrica y otros parámetros sobre la temperatura.
Propiedad física
Nombre de la tecnología de análisis
abreviatura
propiedad física
Nombre de la tecnología de análisis
abreviatura
1. Calidad
1) Termogravimetría
TG
3. Entalpía
9) Calorimetría diferencial de barrido
DSC
2) Medición isobárica del cambio de masa
4. Dimensiones
10) Método de dilatación térmica
3) Detección de escape de gas
EGD
5. 5. Propiedades mecánicas
11) Análisis termomecánico
TMA
4) Análisis de gases de escape
EGA
12) Análisis termomecánico dinámico
DMA
5) Análisis radiotérmico
6. Características acústicas
13) Método termoacústico
6) Análisis térmico de partículas
14) Método termoacústico
2. Temperatura
7) Determinación de la curva de calentamiento
7. Características ópticas
15) Método termoóptico
8) Análisis térmico diferencial
DTA
8. 8. Características eléctricas
16) Método de la termoelectricidad
9. Características magnéticas
17) Método termomagnético
(1) Análisis termogravimétrico (TG)
La termogravimetría (TG) es una técnica para medir el cambio de masa de la muestra con la temperatura o el tiempo bajo el control de una temperatura programada.
Ámbito de aplicación:
(1) Se estudian principalmente los cambios químicos como la estabilidad térmica, la descomposición térmica y la degradación oxidativa de los materiales en gas inerte, aire y oxígeno;
(2) Se estudian todos los procesos físicos que implican un cambio de masa, como la determinación de la humedad, la materia volátil y el residuo, la absorción y la desorción, la tasa de gasificación y el calor de gasificación, la tasa de sublimación y el calor de sublimación, la composición del polímero o la mezcla con el relleno, etc.
Explicación del principio:
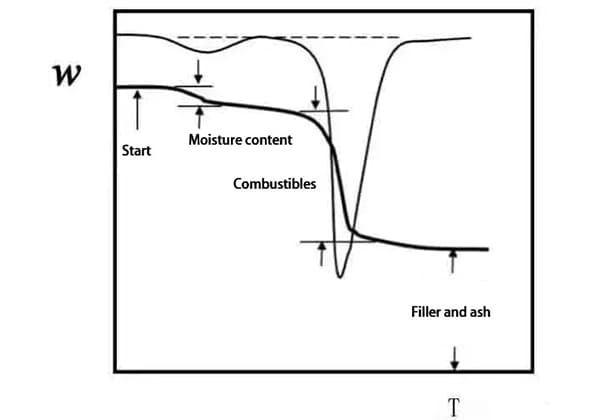
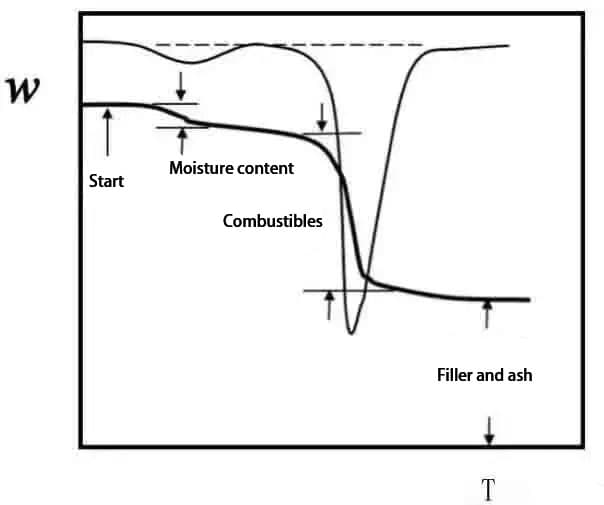
La curva termogravimétrica (curva TG) se obtiene trazando la fracción en peso de la muestra w frente a la temperatura T o el tiempo t: w=f (T o t).
Como la mayor parte del aumento de temperatura es lineal, T y t sólo tienen una constante de diferencia.
La primera derivada de la curva TG con respecto a la temperatura o al tiempo, dw/dT o dw/dt, se denomina curva termogravimétrica diferencial (curva DTG).
En la Fig. 2, el cambio de peso acumulado en Ti en el punto B alcanza el límite inferior de detección de la termobalanza, que se denomina temperatura inicial de reacción;
El cambio de peso en el punto C Tf no puede detectarse, lo que se denomina temperatura de fin de reacción;
Ti o Tf también puede determinarse por extrapolación, que se divide en punto G y punto H;
La temperatura a la que la pérdida de peso alcanza un valor predeterminado (5%, 10%, etc.) también puede tomarse como Ti.
Tp representa la temperatura de máxima tasa de pérdida de peso, correspondiente a la temperatura pico de la curva DTG.
El área del pico es proporcional al cambio de peso de la muestra.
Aplicación práctica:
La termogravimetría se ha convertido en un método importante para estudiar el proceso de cambio térmico de los polímeros debido a su rapidez y sencillez.
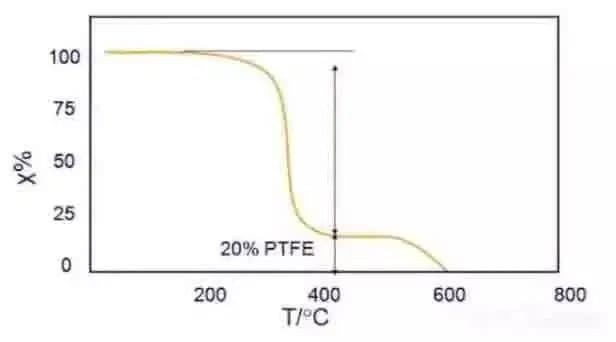
Por ejemplo, la curva TG de la mezcla de PTFE y copolímero acetal de la Fig. 3 puede utilizarse para analizar los componentes de la mezcla.
De la figura se desprende que cuando se calienta en N2, el componente acetal se descompone (alrededor de 80%) a 300~350 ℃, y el PTFE comienza a descomponerse (alrededor de 20%) a 550 ℃.
Factores que influyen:
(a) Velocidad de calentamiento:
Cuanto más rápido aumente la temperatura, mayor será el desfase térmico, mayor será la Ti y Tfy cuanto más amplio sea el intervalo de temperaturas de reacción.
Se sugiere que la muestra de polímero sea de 10K/min, la muestra inorgánica y metálica de 10-20K/min;
(b) Tamaño de las partículas y dosificación de la muestra:
El tamaño de las partículas de la muestra no debe ser demasiado grande y la compacidad del relleno debe ser moderada.
Para un mismo lote de muestras de ensayo, la granulometría y la estanqueidad de cada muestra deberán ser constantes;
(c) Atmósfera:
Las atmósferas más comunes son el aire, el O2, N2He, H2CO2, Cl2 y vapor de agua.
El mecanismo de reacción es diferente en distintas atmósferas. Cuando la atmósfera reacciona con la muestra, la forma de la curva TG se ve afectada;
(d) El material y la forma del plato de muestras.
(2) Análisis termomecánico estático (TMA)
El análisis termomecánico es una técnica para medir la relación funcional entre la deformación de los materiales y el tiempo de temperatura bajo la acción de la temperatura del programa y la carga sin vibración, principalmente midiendo el coeficiente de expansión y la temperatura de transición de fase de los materiales.
Ámbito de aplicación:
El analizador termomecánico estático se utiliza principalmente para el coeficiente de expansión térmica de materiales inorgánicos, materiales metálicos, materiales compuestos y materiales poliméricos (plásticos, caucho, etc.);
Temperatura de transición vítrea;
Punto de fusión;
Punto de reblandecimiento;
Temperatura de deformación térmica de la carga;
Creep, etc.
Aplicación práctica:
(a) Investigación sobre fibra y película:
Puede medir su alargamiento, rendimiento de contracción, módulo y temperatura correspondiente, análisis de deformación por tensión y análisis de tensión en condiciones de congelación y calentamiento;
(b) Caracterización de materiales compuestos:
Además del estudio de la fibra con TMA, el refuerzo de materiales compuestos, la temperatura de transición vítrea TgEl tiempo de gelificación y la fluidez de la resina, el coeficiente de expansión térmica y otras propiedades, así como la estabilidad dimensional y la estabilidad a altas temperaturas de los materiales compuestos multicapa pueden medirse y estudiarse rápidamente con TMA;
(c) Investigación sobre revestimientos:
Es posible saber si el revestimiento se adapta al sustrato y al intervalo de temperatura correspondiente;
(d) Investigación sobre el caucho:
Se puede saber si el caucho sigue teniendo elasticidad y si el tamaño es estable en un entorno de uso duro.
Factores que influyen:
(a) Velocidad de calentamiento:
La distribución de la temperatura de la muestra es desigual cuando la velocidad de calentamiento es demasiado rápida;
(b) Historial térmico de la muestra;
(c) Muestra de defectos:
Porosidad, distribución desigual del relleno, agrietamiento, etc;
(d) Presión aplicada por la sonda:
Generalmente se recomienda 0,001~0,1N;
(e) Cambio químico de la muestra;
(f) Vibración externa;
(g) Calibración:
Calibración de la sonda, temperatura, presión, constante del horno, etc;
(h) Atmósfera;
(i) Forma de la muestra
Si las superficies superior e inferior se aplican en paralelo.
(3) Calorimetría diferencial de barrido (DSC)
Explicación del principio:
La calorimetría diferencial de barrido (DSC) es una tecnología para medir la relación entre la diferencia de potencia entre el material y el material de referencia y la temperatura bajo la temperatura de control del programa.
Existen dos tipos de calorimetría diferencial de barrido: la de compensación y la de flujo térmico.
Se instalan dos grupos de cables calefactores de compensación debajo de los contenedores de muestras y objetos de referencia.
Cuando la diferencia de temperatura ΔT entre la muestra y el objeto de referencia se produce debido al efecto térmico durante el proceso de calentamiento, la corriente que fluye en el cable calefactor de compensación cambiará a través del circuito amplificador térmico diferencial y el amplificador de compensación térmica diferencial.
Cuando la muestra absorbe calor, el amplificador de compensación aumenta inmediatamente la corriente en un lado de la muestra;
Por el contrario, cuando la muestra es exotérmica, la corriente en un lado del material de referencia aumentará hasta que el calor en ambos lados se equilibre y la diferencia de temperatura ΔT desaparezca.
En calorimetría diferencial de barrido, la curva de relación entre el calor aplicado y la temperatura necesaria para mantener la diferencia de temperatura entre la muestra y la referencia en cero por unidad de tiempo es una curva DSC.
El eje vertical de la curva es la cantidad de calentamiento por unidad de tiempo, y el eje horizontal es la temperatura o el tiempo.
El área de la curva es proporcional al cambio de entalpía. En la Fig. 4 se muestra una curva DSC típica.
Ámbito de aplicación:
(1) Determinación de la temperatura de reacción de curado y el efecto térmico de los materiales, como el calor de reacción, la velocidad de reacción, etc;
(2) Determinación de los parámetros termodinámicos y cinéticos de las sustancias, como la capacidad calorífica específica, el calor de transformación, etc;
(3) Determinación de la cristalización, la temperatura de fusión y el efecto térmico de los materiales;
(4) Pureza de la muestra, etc.
Factores que influyen:
(a) Velocidad de calentamiento:
Los resultados reales de las pruebas muestran que una velocidad de calentamiento demasiado alta provocará una distribución desigual de la temperatura en la muestra, y el cuerpo del horno y la muestra también producirán un desequilibrio térmico, por lo que la influencia de la velocidad de calentamiento es muy compleja.
(b) Atmósfera:
Los diferentes gases tienen diferente conductividad térmica, lo que afectará a la resistencia térmica entre la pared del horno y la muestra, y afectará a la temperatura pico y a la entalpía.
(c) Dosificación de la muestra:
No demasiado, para evitar la expansión de la forma del pico y la reducción de la resolución debido a la lenta transferencia de calor interna y al gran gradiente de temperatura.
(d) Tamaño de las partículas de la muestra:
Cuando el tamaño de las partículas de polvo es diferente, debido a la influencia de la transferencia de calor y la difusión, habrá diferencias en los resultados de las pruebas.
(4) Análisis Termomecánico Dinámico (AMD)
El análisis termomecánico dinámico mide la relación entre las propiedades mecánicas de los materiales viscoelásticos y el tiempo, la temperatura o la frecuencia.
La muestra se deforma bajo la acción y el control de una tensión mecánica periódica (sinusoidal).
Ámbito de aplicación:
El analizador termomecánico dinámico se utiliza principalmente para probar la temperatura de transición vítrea, la temperatura de deformación térmica de carga, la fluencia, el módulo de almacenamiento (rigidez), el módulo de pérdida (rendimiento de amortiguación), la relajación de tensiones, etc. de materiales inorgánicos, materiales metálicosmateriales compuestos y materiales poliméricos (plásticos, caucho, etc.).
Principio básico del DMA:
La AMD caracteriza las propiedades de los materiales a través del estado de movimiento molecular.
El movimiento molecular y el estado físico determinan el módulo dinámico (rigidez) y la amortiguación (la energía que pierde la muestra en vibración).
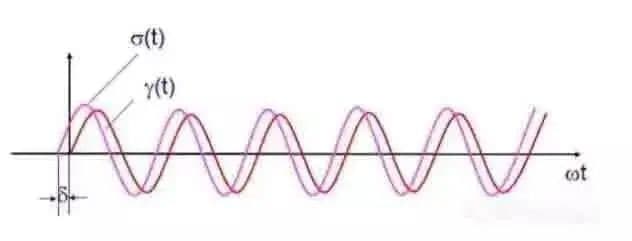
Cuando se aplica a la muestra una tensión alterna sinusoidal de amplitud variable, se generará una deformación sinusoidal de amplitud preseleccionada, y la deformación en las muestras viscoelásticas se retrasará un cierto ángulo de fase δ, como se muestra en la Fig. 5.
La tecnología DMA divide la viscoelasticidad de los materiales en dos módulos:
Módulo de almacenamiento E':
E' está en proporción directa con la elasticidad máxima de la muestra almacenada en cada semana, reflejando los componentes elásticos en la viscoelasticidad del material y caracterizando la rigidez del material;
El módulo de pérdidas E ":
E" es directamente proporcional a la energía consumida por la muestra en forma de calor en cada semana, refleja la parte viscosa de la viscoelasticidad del material y representa la amortiguación del material.
El amortiguamiento del material también se convierte en la fricción interna, expresada como tanδ, y la relación entre la energía perdida por el material en el período semanal y la energía de almacenamiento elástico máximo es igual al módulo de pérdida E "y el módulo de almacenamiento E 'del material.
DMA adopta el barrido de aumento de temperatura, desde la temperatura ambiente auxiliar hasta la temperatura de fusión, tanδ muestra una serie de picos, y cada pico corresponderá a un proceso de relajación específico.
El ángulo de fase tanδ, el módulo de pérdida E "y el módulo de almacenamiento E 'pueden ser medidos por DMA en función de la temperatura, la frecuencia o el tiempo.
No sólo proporciona propiedades mecánicas en una amplia gama de temperaturas y frecuencias, sino que también puede detectar la transición vítrea, la transición a baja temperatura y el proceso de relajación secundaria de los materiales.
Por ejemplo, el pico de pérdida puede representar la transición de cierto movimiento unitario.
La Fig. 6 muestra la curva de poliestireno tgcambiando con la temperatura, de lo que se deduce que el pico puede ser el movimiento del fenilo alrededor de la cadena principal;
El pico es el movimiento del benceno alrededor del enlace que une la cadena principal.
Factores que influyen: velocidad de calentamiento, espesor de la muestra, presencia o ausencia de revestimiento metálico, tipo de fijación, etc.
(5) Análisis dieléctrico dinámico (DETA)
El análisis dieléctrico dinámico es una tecnología para comprobar el cambio de las propiedades dieléctricas de los materiales con la temperatura cuando los materiales se calientan mediante un determinado programa de temperatura controlada bajo un campo eléctrico alterno de una determinada frecuencia.
Principio de análisis dieléctrico:
Los dieléctricos con dipolos se ordenarán direccionalmente con el campo eléctrico externo bajo la acción del campo eléctrico externo.
La polarización del dipolo está relacionada con la temperatura y va acompañada del consumo de energía.
Generalmente, la constante dieléctrica (ε) representa el grado de polarización del dieléctrico bajo el campo eléctrico externo, mientras que la pérdida dieléctrica (D) representa la pérdida de energía causada por el calentamiento de la polarización bajo el campo eléctrico externo.
La disposición direccional de los dipolos bajo la acción del campo eléctrico externo también se recuperará al estado desordenado con la eliminación del campo eléctrico externo.
El tiempo necesario para que el dipolo pase de la disposición regular a la aleatoria se denomina "tiempo de relajación dieléctrica T", según la teoría de Debye:
η es la viscosidad del medio, a es el radio molecular, K es la constante de Boltzmann y T es la temperatura K.
El tiempo de relajación está relacionado con el tamaño y la forma de las moléculas y la viscosidad del medio. Y
Donde: tgδ es la tangente del ángulo de pérdida, y ε0 es la constante dieléctrica bajo campo electrostático; ε∞ es la constante dieléctrica a la frecuencia óptica.
Se puede observar que ε y tgδ son magnitudes físicas relacionadas con el tiempo de relajación τ, y por tanto relacionadas con la estructura molecular, el tamaño y la viscosidad del medio, que es la base para utilizar las propiedades dieléctricas para estudiar la estructura molecular de las sustancias.
Se puede demostrar a partir de las dos ecuaciones anteriores que cuando:
Cuando, ε 'tiene un valor máximo, f0 se denomina "frecuencia de polarización".
Es decir, cuando la frecuencia del campo eléctrico externo es la frecuencia de polarización, la pérdida dieléctrica es muy grande.
Ámbito de aplicación:
Esta técnica se ha utilizado ampliamente para estudiar la estructura molecular, el grado de polimerización y el mecanismo polimérico de los materiales dieléctricos.
En cuanto a los objetos de aplicación, existen resinas termoplásticas y termoendurecibles como el éster metílico de poliacrilato, el cloruro de polivinilo, la poliamida, la poliimida, el poliestireno, el fenol formaldehído, el epoxi y la cera.
Además, hay polifenil arce y polibenzimidazol en resina resistente a altas temperaturas, y proteínas en compuestos biológicos.
Sus aplicaciones específicas también incluyen plásticos reforzados, materiales de moldeo, revestimientos, adhesivos, caucho, vidrio, cerámica y otros óxidos metálicos.
En el laboratorio, la DETA puede utilizarse como una potente herramienta para la investigación viscoelástica, como los ensayos de propiedades mecánicas dinámicas y termomecánicas.
En la producción industrial, puede utilizarse en la fabricación de resinas, el control de calidad, el precurado y el control del grado de curado.
Como fundador de MachineMFG, he dedicado más de una década de mi carrera a la industria metalúrgica. Mi amplia experiencia me ha permitido convertirme en un experto en los campos de la fabricación de chapa metálica, mecanizado, ingeniería mecánica y máquinas herramienta para metales. Estoy constantemente pensando, leyendo y escribiendo sobre estos temas, esforzándome constantemente por mantenerme a la vanguardia de mi campo. Deje que mis conocimientos y experiencia sean un activo para su empresa.
Imagine desbloquear la precisión de las máquinas herramienta con una simple superficie de cristal. Las escalas de rejilla lineal hacen precisamente eso, transformar líneas finas en mediciones de gran precisión. Este artículo explora cómo estas...
¿Alguna vez se ha preguntado cómo se suavizan los bordes afilados de las piezas metálicas? Este proceso, conocido como biselado, transforma las esquinas peligrosas y dentadas en superficies angulosas más seguras. En este artículo...
¿Alguna vez se ha preguntado qué marcas de rodamientos son las mejores del mundo? En esta entrada del blog, exploraremos los principales fabricantes de rodamientos conocidos por su excepcional calidad, innovación y...
¿Se ha preguntado alguna vez quién mueve el mundo entre bastidores? En esta entrada del blog, nos adentraremos en los principales fabricantes de generadores que mantienen la luz encendida...
¿Es usted un aspirante a ingeniero mecánico que busca sobresalir en su campo? En esta entrada del blog, exploraremos los 10 programas de diseño de ingeniería mecánica más...
¿Alguna vez se ha preguntado cómo se garantiza la integridad de las tuercas y tornillos soldados de su coche? Este artículo revela el meticuloso proceso de comprobaciones e inspecciones de calidad que mantienen su vehículo...
¿Se ha preguntado alguna vez qué hace que un motor eléctrico funcione sin sobrecalentarse? Conocer las temperaturas de funcionamiento seguras de los motores es crucial para su longevidad y rendimiento. En este artículo,...
¿Alguna vez se ha preguntado por el fascinante mundo de la fundición? Este antiguo proceso de fabricación, en constante evolución, da forma a nuestra vida cotidiana de innumerables maneras. En esta entrada del blog, exploraremos la...
¿Alguna vez se ha preguntado cómo funcionan los engranajes de su coche o su avión? Este artículo desvela los principales fabricantes de engranajes que están dando forma al futuro de la ingeniería mecánica. Aprenderá...